














DAATNEWS PORTUGAL Julho 2025


Será que o nosso ADN também nos rouba o sono?
Autor do comentário: Dra. Catarina Guimarães. MD, Pneumologista, ULS Alto Ave.
Lucas Møller Larsen, Sine Voss Winther, Asbjørn Kørvel-Hanquist, Sarah C W Marott, Eskild M Landt, Preben Homøe, Børge G Nordestgaard¡, Morten Dahl.


O estudo apresentado trata-se de uma investigação sobre a possível associação entre a deficiência de α1-antitripsina (DAAT) e a apneia do sono (AS), incluindo a apneia obstrutiva do sono (AOS), com base numa coorte nacional dinamarquesa. A utilização de uma amostra populacional extensa (n = 29.452), com seguimento prolongado (mediana de 62 anos) e a utilização de dados de registo nacionais com cobertura quase universal, representa uma clara mais-valia metodológica, reduzindo vieses de seleção e minimizando perdas de seguimento.
Foi realizada a estratificação por múltiplos fatores confundidores relevantes, como DPOC, doenças cardiovasculares, diabetes tipo 2, doença hepática, idade e sexo o que permite um controlo mais rigoroso das associações observadas. A principal conclusão é que os indivíduos com DAAT têm um risco aumentado de AS (HR 1,81; IC 95 %: 1,36–2,40), independentemente da presença de DPOC (Fig.1). Resultados semelhantes foram encontrados quando avaliado o risco para AOS mas sem resultados estatisticamente significativos (HR 1.47 (IC 95 %: 0,95–2,28) (Fig.2). Estes dados são particularmente relevantes, pois sugere uma possível via fisiopatologia distinta, sugerindo um mecanismo adicional à limitação ventilatória periférica, centrado na colapsabilidade faríngea e possivelmente relacionada com a degradação de tecidos elásticos na via aérea superior.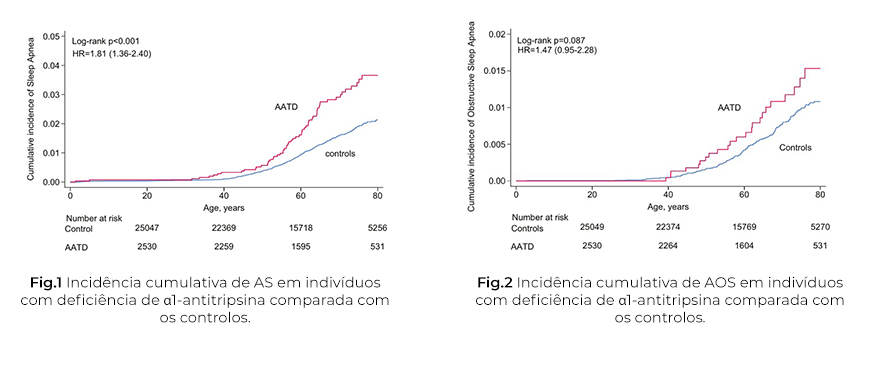 No entanto, existem algumas limitações metodológicas. A definição de AS e AOS baseou-se exclusivamente em códigos ICD-10, o que pode comprometer a sensibilidade e especificidade diagnóstica. A ausência de dados polissonográficos, como o índice de apneia-hipopneia (IAH), impede a caracterização fenotípica dos casos e a estratificação por gravidade. Esta limitação é crítica, sobretudo tendo em conta a elevada taxa de subdiagnóstico da AS na população geral, o que pode enviesar os riscos estimados.
No entanto, existem algumas limitações metodológicas. A definição de AS e AOS baseou-se exclusivamente em códigos ICD-10, o que pode comprometer a sensibilidade e especificidade diagnóstica. A ausência de dados polissonográficos, como o índice de apneia-hipopneia (IAH), impede a caracterização fenotípica dos casos e a estratificação por gravidade. Esta limitação é crítica, sobretudo tendo em conta a elevada taxa de subdiagnóstico da AS na população geral, o que pode enviesar os riscos estimados.
A discrepância entre os resultados para AS (estatisticamente significativos) e para AOS (não significativos) levanta questões quanto à consistência da codificação. Embora os autores justifiquem esta diferença com base na codificação incompleta, esta limitação fragiliza a validade das conclusões específicas para AOS. Importa ainda considerar o potencial viés de deteção: indivíduos com DAAT podem ter maior contacto com o sistema de saúde, o que poderia conduzir a uma maior probabilidade de diagnóstico de SA.
Em suma, trata-se de um estudo com metodologia sólida e implicações clínicas relevantes, sugerindo que a DAAT pode constituir um fator de risco independente para AS. No entanto, estudos com caracterização clínica detalhada e confirmação fisiológica do diagnóstico são necessários para validar estes achados.


Novas guidelines sobre deficiência de alfa 1 antitripsina – o que preconizam os Canadianos?
Autor do comentário: Dra. Joana Gomes. MD, Pneumologista, ULS de Santo António.
Paul Hernandez, Yohan Bossé, Pam Bush, Kenneth R Chapman, François Maltais, Erika D Penz, Brandie L Walker, Avtar Lal, Darcy D Marciniuk.
Meta-Analysis. Chest. 2025 Apr;167(4):1044-1063. doi: 10.1016/j.chest.2024.08.037. (Abstract do artigo)
Tendo em conta os estudos e meta-análises nas áreas de diagnóstico e tratamento da deficiência de alfa 1 antitripsina (DAAT), a Canadian Thoracic Society (CTS) decidiu emitir uma guideline prática focada apenas na doença respiratória associada a DAAT. Neste âmbito, a população alvo definida são os indivíduos com doença pulmonar obstrutiva crónica (DPOC) e níveis de alfa 1 antitripsina (AAT) < 11umol/L ou < 0,57 g/L, considerando que estes serão os principais candidatos a terapêutica de substituição com AAT.
Em relação ao diagnóstico, a CTS preconiza testar todos os indivíduos com DPOC aquando do diagnóstico, indivíduos com asma de início em idade adulta com obstrução persistente e indivíduos com bronquiectasias inexplicadas. Existindo uma elevada suspeição clínica de DAAT (DPOC com início antes dos 40 anos de idade, DPOC em indivíduos com carga tabágica < 10 UMA, enfisema basal panlobular, história familiar de DPOC ou DAAT e antecedentes de icterícia neonatal) sugerem medição dos níveis séricos de A1AT e sequenciação do DNA do gene SERPINA1. No caso de suspeita clínica moderada (DPOC, bronquiectasias inexplicadas, asma de início em idade adulta com obstrução persistente, cirrose hepática, paniculite e vasculite associada a granulomatose com poliangeíte) realizar medição dos níveis séricos de AAT seguido de sequenciação do gene SERPINA 1 se AAT < 23umol/L ou 1,2g/L. A recomendação do uso da sequenciação do DNA do gene SERPINA 1 prende-se com o facto de ser o gold standard e permitir aferir corretamente não só os alelos graves, como alelos nulos ou variantes raras. Permite também clarificar situações de défice intermédio de AAT. O rastreio genético está recomendado aos parentes de 1º grau (pais, irmão e filhos) do caso índice, quer este seja homozigoto ou heterozigoto.
Quanto à terapêutica de substituição com AAT, esta sociedade define como objetivos preservar a densidade pulmonar medida por TC tórax e reduzir mortalidade. A terapêutica de substituição com AAT é recomendada nesta guideline em doentes com DPOC e todos os seguintes parâmetros: não-fumador ou ex-fumador (> 6 meses); FEV1 < 80 %; enfisema pulmonar documentado por TC; genótipo SERPINA 1 associado com DAAT; níveis séricos de AAT gravemente reduzidos (< 11 umol/L ou < 0,57 g/L); encontrarem-se sob terapêuticas farmacológica e não-farmacológica otimizadas para a DPOC. Além destes, na discussão destas orientações são referidos ainda como critérios a indicação do benefício desta terapêutica por médico pneumologista e a exclusão de doentes que foram submetidos a transplante pulmonar.
A CTS refere ainda algumas necessidades futuras em termos de investigação e evidência, tais como novas formas mais práticas de administração da terapêutica de substituição (inalada ou oral); possibilidade de tratar doentes com enfisema pulmonar com função pulmonar preservada, assim como doentes sem enfisema e com outros fenótipos clínicos (asma, bronquiectasias); terapia genética; e estudos de larga escala longitudinais com coortes e populacionais que permitam compreender a progressão desta doença a longo prazo.

Raros, mas não irrelevantes: Genótipos raros e défice grave no Défice de Alfa-1-Antitripsina
Autor do comentário: Dra. Maria João Silva. MD, Pneumologista. Unidade Local de Saúde Região Leiria.
Ilaria Ferrarotti, Davide Piloni, Asia Filosa, Stefania Ottaviani, Valentina Barzon, Alice Maria Balderacchi, Luciano Corda, Christine Seebacher, Sara Magni, Francesca Mariani, Paolo Baderna, Paola Confalonieri, Leonardo Iannacci, Silvia Mancinelli, Paola Putignano, Carlo Albera, Giulia Maria Stella, Maria Cristina Monti, Angelo Guido Corsico.
Pulmonology. 2025 Dec 31;31(1):2429911. doi: 10.1080/25310429.2024.2429911. (Abstract do artigo)
O défice de Alfa-1-Antitripsina (DAAT) é uma condição autossómica codominante que traduz risco aumentado de doença pulmonar e hepática.1 As formas graves implicam que ambos os alelos sejam patogénicos, traduzindo baixos níveis plasmáticos de proteína funcional, ausência completa desta ou uma proteína disfuncional.2
Cerca de 95 % dos casos graves apresentam genótipo PI*ZZ associado a mais patologia hepática e pulmonar e, como tal, a maioria dos estudos incide sobre este.3
As variantes raras, com uma prevalência inferior a 1 %, podem estar associadas a alelos disfuncionais ou nulos, caracterizadas por níveis plasmáticos reduzidos, acumulação intra-hepática e doença pulmonar.4 Alelos nulos estão associados a proteína não detetável na circulação e em homozigotia apresentam doença pulmonar mais grave que indivíduos PI*ZZ.2
O artigo caracteriza indivíduos com genótipos raros comparando-os com outros de genótipos mais frequentes e com doença grave (PI*ZZ), sendo que estes representam 23,3 % dos indivíduos com DAAT grave no Registo Italiano, a maior frequência mundial entre registos.5
Relativamente às suas características demográficas, exposições, idade início de sintomas estas não eram distintas dos doentes com genótipo PI*ZZ. A distribuição do fenótipo pulmonar foi semelhante entre os dois grupos com o enfisema como patologia pulmonar mais prevalente e funcionalmente ambos apresentavam mais obstrução pulmonar. Uma outra semelhança são os valores de AAT (Alfa-1-Antitripsina) que estavam abaixo do limiar de proteção.
O trabalho vem assim afirmar que doentes com genótipos raros, quando apresentam défice grave, são muito semelhantes aos pacientes com genótipo PI*ZZ.
No entanto, dada a sua baixa frequência, o impacto clínico destes permanece pouco estudado. São necessários melhores métodos de diagnóstico,6 com aplicação de algoritmos validados, para deteção de novas variantes e caracterização do mecanismo patogénico de cada variante, nomeadamente, pesquisas sobre a capacidade de polimerização e avaliação de polímeros circulantes.4
Como consequência da falta de dados, a terapêutica de substituição não está totalmente estudada nestes doentes. No entanto, dada a similaridade de comportamento destes com doentes PI*ZZ, podemos questionar se estes não deveriam ser elegíveis para terapêutica.
Registos nacionais e internacionais, como o European Alpha-1 Research Collaboration (EARCO),7 são fundamentais para melhor compreender a história natural da doença e o impacto das terapias, sendo por isso fundamental a inclusão destes para que futuramente possamos oferecer as mesmas intervenções preventivas e terapêuticas.
Bibliografia:
- Greene CM, Marciniak SJ, Teckman J, Ferrarotti I, Brantly ML, Lomas DA, et al. α1-Antitrypsin deficiency. Nat Rev DisPrimers. 2016;2(1):16051. doi:10.1038/nrdp.2016.51.
- Ferrarotti I, Carroll TP, Ottaviani S, Fra AM, O’Brien G, Molloy K, et al. Identification and characterisation of eightnovel SERPINA1 null mutations. Orphanet J Rare Dis. 2014;9(1):172. doi:10.1186/s13023-014-0172-y.
- Strnad P, McElvaney NG, Lomas DA, Longo DL. Alpha1-Antitrypsin deficiency. N Engl J Med. 2020;382(15):1443–1455. doi:10.1056/NEJMra1910234.
- Ottaviani S, Bartoli G, Carroll TP, Gangemi F, Balderacchi AM, Barzon V, et al. Comprehensive clinical diagnosticpipelines reveal new variants in alpha-1-antitrypsin deficiency. Am J Respir Cell Mol Biol. 2023;69(3):355–366. doi:10.1165/rcmb.2022-0470OC.
- Meira L, Boaventura R, Seixas S, Sucena M. Alpha-1 antitrypsin deficiency detection in a Portuguese population.COPD J Chronic Obstr Pulm Dis. 2018;15(1):4–9. doi:10.1080/15412555.2017.1414779.
- Ottaviani S, Barzon V, Buxens A, Gorrini M, Larruskain A, El Hamss R, et al. Molecular diagnosis of alpha1-antitrypsindeficiency: a new method based on Luminex technology. J Clin Lab Anal. 2020;34(7):e23279. doi:10.1002/jcla.23279.
- Greulich T, Altraja A, Barrecheguren M, Bals R, Chlumsky J, Chorostowska-Wynimko J, et al. Protocol for the EARCOregistry: a pan-European observational study in patients with α1-antitrypsin deficiency. ERJ Open Res. 2020;6(1):00181–2019. doi:10.1183/23120541.00181-2019.PULMONOLOGY 11.

Desmistificar o PI*Mmalton: nova luz sobre uma mutação rara do Défice de Alfa-1-Antitripsina
Autor do comentário: Dra. Beatriz Ferraz. MD, Interna de Pneumologia. Unidade Local de Saúde de Santo António
Beatriz D Ferraz, Maria Sucena, Margarida Fonseca Cardoso, Alice M Turner, José María Hernández-Pérez, María Torres-Duran, Hanan Tanash, Carlota Rodríguez-García, Jens-Ulrik Jensen, Angelo Corsico, José Luis López-Campos, Kenneth Chapman, Christian F Clarenbach, Joana Gomes, Marc Miravitlles, Beatriz Lara.
BMC Pulm Med. 2025 Apr 23;25(1):187. doi: 10.1186/s12890-025-03651-8. (Abstract do artigo)
Este estudo apresenta a maior caracterização, até à data, de indivíduos portadores do alelo PI*Mmalton com défice de alfa-1 antitripsina (DAAT), utilizando os dados do registo internacional EARCO.
Os doentes foram estratificados em dois subgrupos clinicamente relevantes — deficência moderada (PI*Mmalton/S ou PI*Mmalton/I) e grave (PI*Mmalton/Z, PI*Mmalton/Mmalton e outras combinações com variantes severas) — e foram comparados com a coorte de indivíduos com o genótipo PI*ZZ.
Os resultados mostraram que os indivíduos com genótipo PI*Mmalton graves apresentam uma função pulmonar comprometida e níveis de AAT semelhantes aos observados no grupo PI*ZZ, com reduções marcadas do rácio FEV₁/FVC e prevalência elevada de enfisema. Estes achados reforçam o potencial patogénico da mutação PI*Mmalton quando combinada com outro alelo grave, defendendo o seu reconhecimento clínico como genótipo de alto risco.
Por outro lado, o grupo PI*Mmalton moderado evidenciou menor compromisso respiratório e níveis séricos de AAT mais elevados, o que confirma que nem todos os portadores de PI*Mmalton partilham o mesmo grau de risco e que o fenótipo depende fortemente do alelo concomitante. Importa também referir que as manifestações hepáticas foram comparáveis entre todos os grupos, sugerindo que o risco hepático poderá justificar uma vigilância semelhante independentemente da gravidade da deficiência.
O estudo evidencia ainda uma distribuição geográfica com os portadores da variante PI*Mmalton sobretudo nos países mediterrânicos, nomeadamente Espanha, Portugal e Itália, o que vem corroborar dados prévios e reforça a importância de estratégias de rastreio baseadas na epidemiologia genética regional.
Um ponto forte deste trabalho é a utilização de dados do mundo real, recolhidos de forma multicêntrica e padronizada segundo o protocolo do EARCO. No entanto, a natureza transversal da análise limita a avaliação da progressão da doença, pelo que se sugere o acompanhamento longitudinal para aprofundar o conhecimento sobre o prognóstico desta variante rara.
Em suma, este estudo representa um avanço significativo na compreensão da DAAT associada ao alelo PI*Mmalton. Defende uma abordagem clínica diferenciada com base no genótipo e sugere que os indivíduos com formas graves de PI*Mmalton devem ser geridos de forma semelhante aos doentes PI*ZZ. Estes resultados sustentam ainda a inclusão destes doentes em ensaios clínicos futuros e reforçam a necessidade de estratégias terapêuticas adaptadas a genótipos raros de DAAT.
Coordenação Científica
Dra. Joana Maria Lobo Gomes
MD, Pneumologista,
ULS de Santo António.
Dra. Catarina Sofia Romano Gonçalves Guimarães
MD, Pneumologista,
ULS Alto Ave.
Apoio
Dra. Rosa Leal
Medical Scientific Liaison
CSL™
Comentadores
Dra. Maria João Silva
MD, Pneumologista
Unidade Local de Saúde Região Leiria
Dra. Beatriz Ferraz
MD, Interna de Pneumologia
Unidade Local de Saúde de Santo António

